


Current News
Looking for a topic for your Bachelor-/Master-thesis or project work?
Check here for topics for Bachelor-theses or project works.
Check here for topics for Master-theses.
Check here for open PhD positions.
Or contact Gerhard Kahl for topics/open positions.
Open PhD position: Soft Matter Systems under non-equilibrium conditions
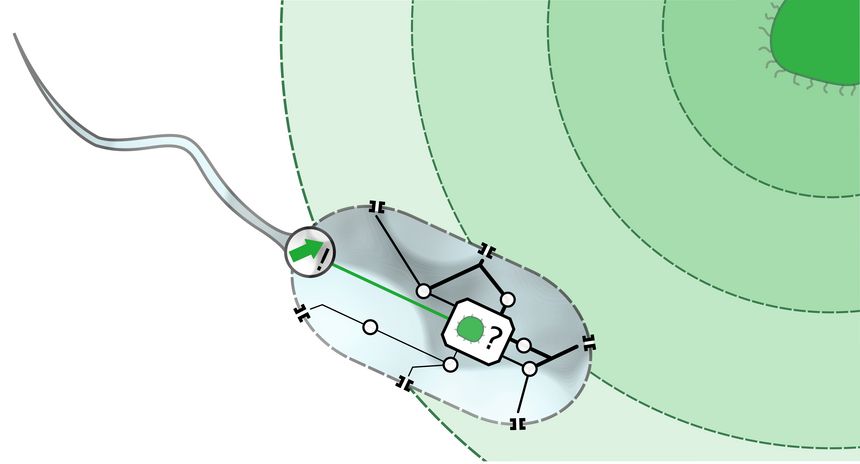
Soft matter systems are ubiquitous in our daily lives, with examples ranging from biology to technological applications. This project offers the possibility to work on bio-related soft matter systems under non-equilibrium conditions, i.e., in a highly actual and strongly interdisciplinary research field. To be more specific we will use - similar as in experiment - external fields (such as fluid flow and external forces) to induce these conditions (see figure).
We warmly welcome a highly motivated and enthusiastic new group member, with experience and skills in computational sciences.
Please find more details on the newly opened PhD-position [here].
How to reach your desired destination as a unicellular organism
How do simple creatures autonomously move towards certain locations?
Artificial intelligence and a proper physical model developed at the TU Wien can explain this.
It is still an unresolved question how simple organisms without a distinct nervous system can make decisions such as "Which way to go?" and "How to navigate there?".
Unicellular organisms can accomplish precisely these tasks seemingly effortlessly: Swimming microorganisms and migrating cells have developed various strategies in order to move in nutrient-rich or other chemical environments.
We apply a genetic algorithm to the internal decision-making machinery of a model microswimmer and show how it learns to approach nutrients in static and dynamic environments. Strikingly, the emerging dynamics resemble the well-known run-and-tumble motion of swimming cells. We demonstrate that complex locomotion and navigation strategies in chemical environments can be achieved by developing a surprisingly simple internal machinery, which in our case, is represented by a small artificial neural network.
Our findings, published in PNAS, shed light on how small organisms have developed the capability to conduct environment-dependent tasks: B. Hartl, M. Hübl, G. Kahl, A. Zöttl: Microswimmers learning chemotaxis with genetic algorithms, PNAS 2021, 118 (19) e2019683118.
Computer simulations explain why bacteria can swim faster in biofluids than in water
Bacteria in the human body typically move through complex, biopolymeric fluids. Counterintuitively, it has been shown experimentally that bacteria can move faster in polymeric fluids than in water, although the viscosity is higher in such fluids. Using extensive computer simulations Andreas Zöttl demonstrated together with Julia M. Yeomans from Oxford University that a non-uniform distribution of polymeric material around the bacterium leads to an apparent slip velocity close to the bacterium which reduces the effective friction around it. They also developed a theoretical framework which predicts under which conditions a swimming speedup can be expected.

Published in Nature Physics:
A. Zöttl, J. M. Yeomans: Enhanced bacterial swimming speeds in macromolecular polymer solutions, Nat. Phys. 15, 554 (2019).
Nanocages in the lab and in the computer
How to create nanocages, i.e., robust and stable objects with regular voids and tunable properties? Short segments of DNA molecules are perfect candidates for the controllable design of novel complex structures. Clemens Jochum and Gerhard Kahl from our group and Nataša Adžić and Christos Likos from the University of Vienna along with experimentalists from Foschungszentrum Jülich and Cornell University investigated methodologies to synthesize DNA-based dendrimers in the lab and to predict their behaviour using detailed computer simulations. The results were published in RSC Nanoscale. This research project is supported by the Austrian Science Fund (FWF) and by the Deutsche Forschungsgemeinschaft (DFG). The computational calculations were in part performed on the Vienna Scientific Cluster (VSC).
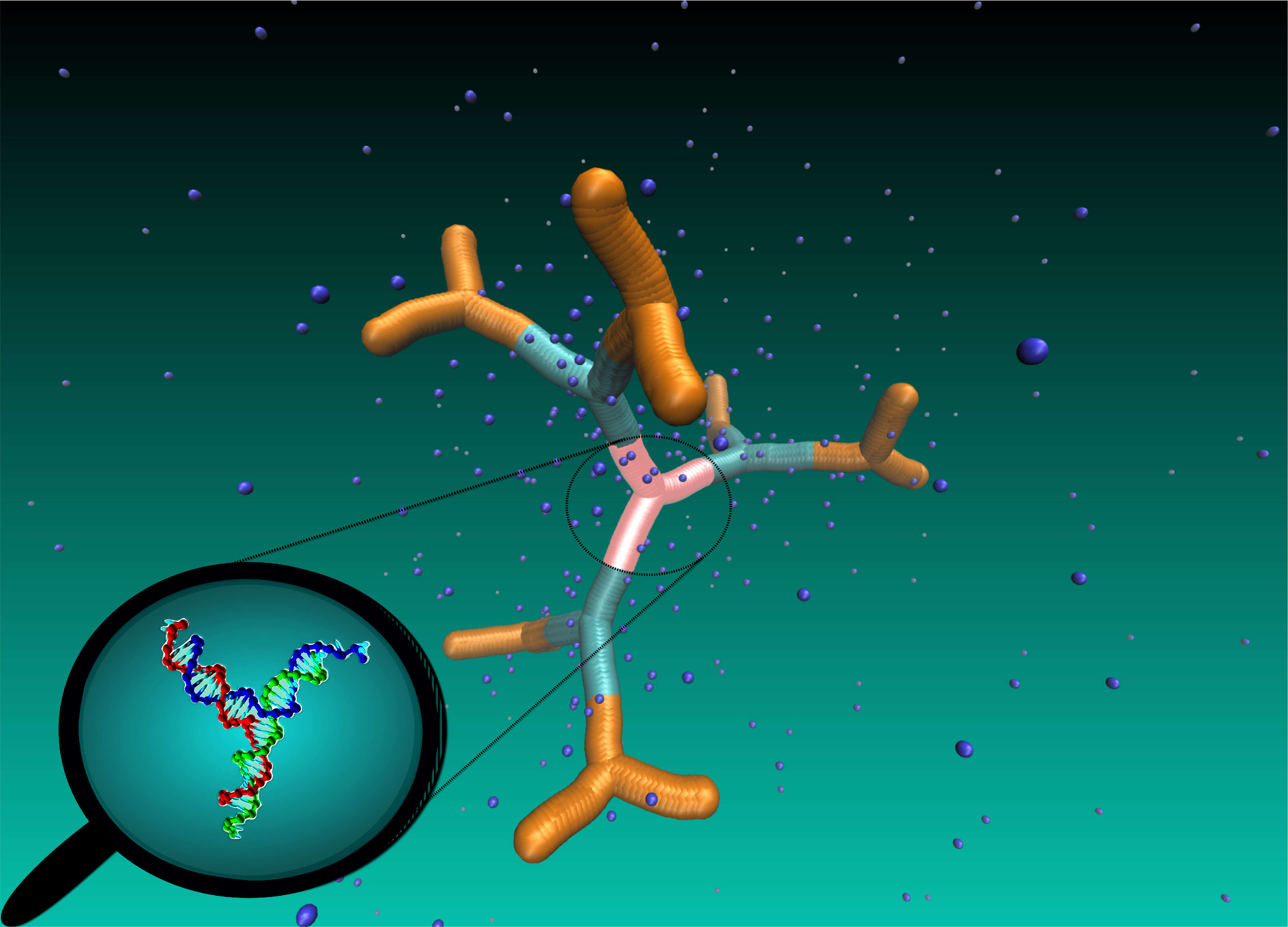
Featured in university newsletters: University of Vienna, TU Wien (English), TU Wien (German)
Press coverage: ORF (German), Der Standard (German), APA (German), Phys.org (English),
Published and featured as a backcover article in RSC Nanoscale:
C. Jochum, N. Adžić, E. Stiakakis, T. L. Derrien, D Luo, G. Kahl, C. N. Likos: Structure and stimuli-responsiveness of all-DNA dendrimers: theory and experiment, Nanoscale, 11, 1604-1617 (2019).
Emanuela Bianchi awarded START grant 2018
We are very happy and proud to announce that Emanuela Bianchi was awarded a START grant by the FWF for her research proposal "Heterogeneously Charged Colloids for Materials Design". The START Programme targets outstanding young researchers of any discipline and aims to provide researchers with long-term and extensive financial security to plan their research and to build up their own research groups thereby qualifying themselves for senior research positions. Every year only six researchers, who need to provide an excellent research proposal and an exceptional international track record, are awarded this prestigious prize, which comes with funding from 800.000 € up to 1.200.000 €. We wish Emanuela all the best with her research project.
Featured in the newsletter by TU Wien: Drei START-Preise für die TU Wien
Update: See Emanuela's group homepage for more information

Emanuela Bianchi (letft) is one of the three recipients of the 2018 START grants at TU Wien.
Contact Information
Institut für Theoretische Physik
Technische Universität Wien
Wiedner Hauptstrasse 8-10/136
A-1040 Wien, Austria
Tel : +43 1 5880113632
Fax : +43 1 5880113699
E-mail : gkahl(at)tph.tuwien.ac.at